
Welttag der Hämophilie
Im Jahr 1963 gründete der kanadische Geschäftsmann Frank Schnabel die World Federation of Hemophilia (WFH) und auf Frank Schnabel ist auch der jährliche Welttag der Hämophilie am 17.04. zurückzuführen, da dieser an diesem Tag geboren worden ist. Also der perfekte Tag, sich mit der doch noch ziemlich unbekannten Erkrankung zu beschäftigen.
Was ist die Hämophilie?
Der Begriff Hämophilie stammt aus dem Altgriechischen (haima = „Blut“ & philia = „Neigung“) und beschreibt eine Erbkrankheit, welche eine Störung der Blutgerinnung zum Ergebnis hat. Bei einer Hämophilie gerinnt das Blut der Betroffenen nicht oder nur verzögert und es kann je nach Ausprägung auch zu spontanen Blutungen ohne sichtbare Verletzungen o.Ä. kommen, v.a. auch innere Blutungen. Wichtig ist es hier aber zu betonen, dass z.B. Schnittverletzungen oder Schürfungen in den meisten Fällen keinen höheren Blutverlust bedeuten als bei nicht von Hämophilie betroffenen Menschen, aber durch die verzögerte Blutgerinnung sorgt dafür, dass der Schorf im Wundbereich immer wieder aufbricht und so die Blutung abhängig von der Hämophilie-Schwere nur sehr langsam oder gar nicht zu stillen ist.
Die Hämophilie ist ist schon seit Jahrhunderten bekannt, so finden sich erste Erwähnungen im Rahmen von Beschneidungen schon im 5. Jahrhundert im Talmud. Erstmals genauer beschrieben wurde die Hämophilie als Erbkrankheit 1803 vom US-amerikanischen Arzt John Conrad Otto. Durch die Geschichte hindurch ist die Hämophilie auch bekannt als „Krankheit der Könige“, weil diese in den europäischen Königshäusern gehäuft auftrat und wahrscheinlich auf Königin Victoria von Großbritannien zurückzuführen ist.
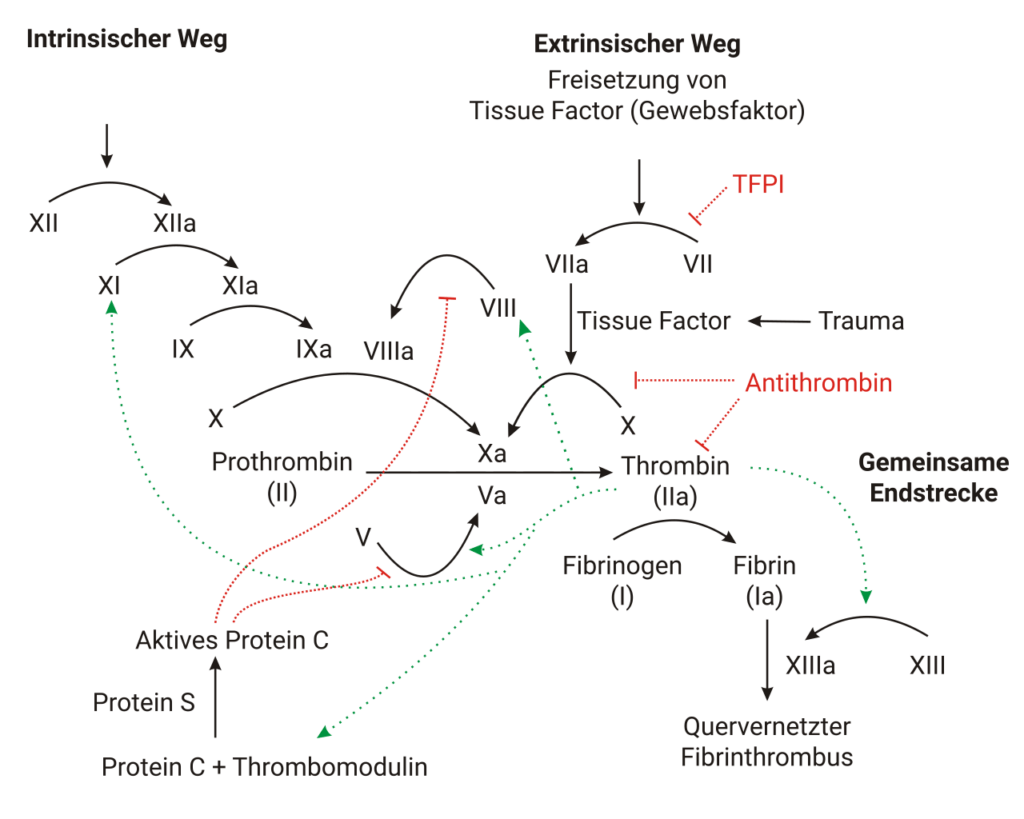
Um die Ursache für die Hämophilie genauer zu verstehen ist es nötig kurz tiefer in das Thema Blutgerinnung einzusteigen. Damit die Gerinnungskaskade funktioniert, müssen die Gerinnungsfaktoren in ausreichender Menge im Blut vorliegen. Da die Gerinnungskaskade ein hochkomplexer Prozess ist, reicht es schon aus, dass ein Gerinnungsfaktor nicht vorliegt oder nicht ordentlich funktioniert und so der ganze Mechanismus nicht vollständig abläuft. Bei der Hämophilie sitzt das Problem nicht am Anfang der Gerinnungskaskade, die zwei ersten Phasen, also vorläufiger Gefäßverschluss & Blutgerinnung bzw. Blutpfropfbildung, laufen normal ab. Das Problem tritt erst in der 3. Phase, der Fibrinolyse/Wundverschluss, auf. Grund hierfür ist das fehlende Fibrin, bedingt durch einen Mangel best. Gerinnungsfaktoren. Kurz und knapp gesagt ist die Hämophilie ein Mangel an Gerinnungsfaktoren. Da aber nicht alle Gerinnungsfaktoren davon betroffen sind bzw. fehlen, sondern i.d.R. nur zwei, unterscheidet man die Hämophilie primär in die Hämophilie A und Hämophilie B. Bei der Hämophilie A fehlt der Gerinnungsfaktor VIII und bei der Hämophilie B der Gerinnungsfaktor IX.
{kind=link}
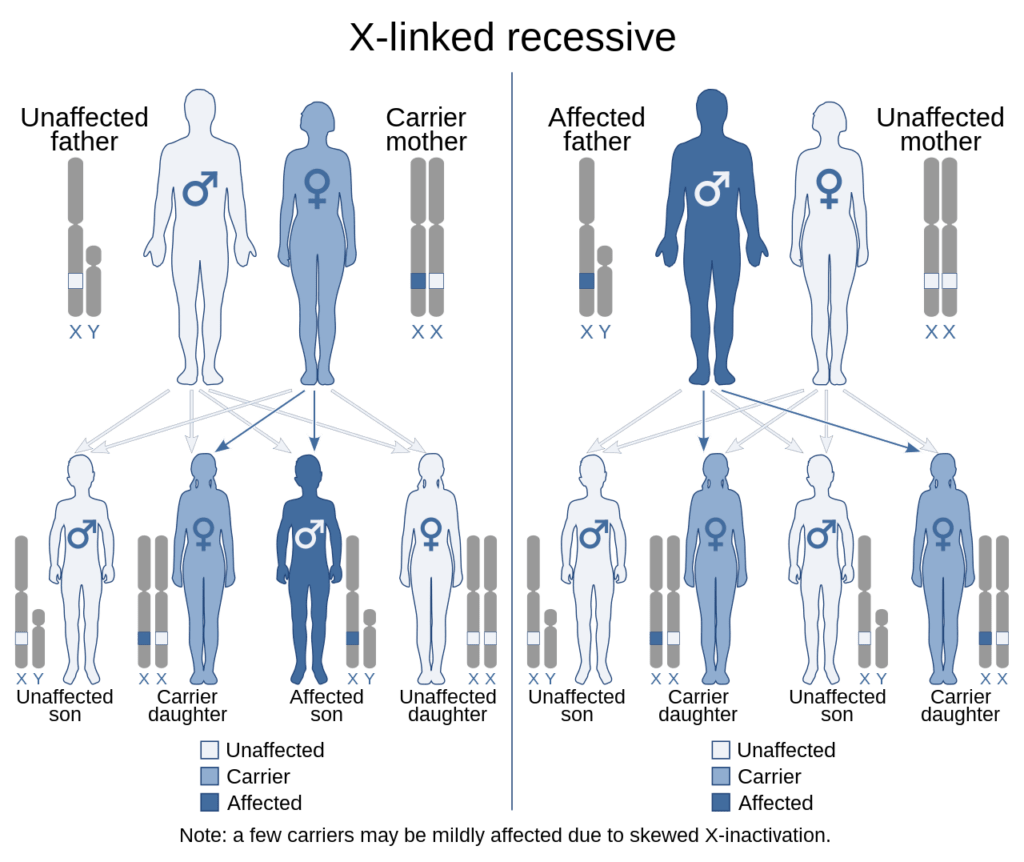
Wie schon erwähnt handelt es sich bei der Hämophilie um eine Erbkrankheit, also eine Erkrankung die vererbt wird. Die Vererbung der Hämophilie nach dem Schema des nachfolgenden Bildes. Die Grafik erklärt auch wieso von der Hämophilie hauptsächlich Männer betroffen sind. Der Grund ist hier die Veränderung auf dem X-Chromosom, welches klassisch, rezessiv vererbt wird und zu einer Mutation im F8- oder F9-Gen führt. Bei Frauen tritt die Hämophilie meist nicht auf, da auch wenn ein X-Chromosom betroffen ist, das andere X-Chromosom dies kompensieren kann.
.svg){kind=link}
Zum Schluss sollte aber betont sein, dass die Gefahr hier bei uns zu verbluten in der heutigen Zeit eigentlich nicht mehr möglich. Grund hierfür sind die guten Therapiemöglichkeiten in hoch entwickelten Ländern wie Deutschland. Hier bei uns geht man sogar schon fast von der vollständigen Angleichung der Lebenserwartung hämophiler Patient*innen im Vergleich zur gesunden Normalbevölkerung aus.
Zahlen & Fakten
- weltweit > 200.000 – 400.000 Menschen mit irgendeiner diagnostizierten Form von Hämophilie (Schätzungen gehen von > 1.100.000 Menschen mit Hämophilie aus)
- in Deutschland leben etwa 6.000 Hämophilie-Patient*innen (50 % haben Hämophilie A)
- ca. 80 % der Hämophilie-Patient*innen hat eine Hämophilie A (Faktor-VIII-Mangel)
- Hämophilie A-Häufigkeit bei Männern: 1 von 5.000
- Hämophilie B-Häufigkeit bei Männern: 1 von 25.000 – 30.000
- ca. 1/3 der Neugeborenen werden in Familien geboren, bei denen keine Hämophilie bekannt ist
- Blutungsneigung ist bei Hämophilie B insgesamt etwas geringer
- erste Hinweis auf Hämophilie gibt es oftmals im Alter zw. 12. – 18. Lebensmonat, wenn die Kindern anfangen zu krabbeln/laufen
- durchschnittliches Diagnosealter in den USA
- 36 Monate bei leichter Hämophilie
- 8 Monate bei mittlerer Hämophilie
- 1 Monat bei schwerer Hämophilie
- Schweregradverteilung am Beispiel der USA (ca. 30.000 – 33.000 Betroffene)
- 60 % schwere Hämophilie
- 15 % moderate Hämophilie
- 25 % milde Hämophilie
- Mortalitätsrate sowie Anzahl KH-Aufenthalte ca. 40 % niedriger bei Behandlung in spezialisiertem Zentrum
- Prophylaxe senkt ICB-Risiko um ca. 50 %
Hämophilie-Formen
- Hämophilie A (Faktor-VIII-Mangel)
- Hämophilie B (Faktor-IX-Mangel)
- Hämophilie C bzw. Rosenthal-Syndrom (Faktor-XI-Mangel)
- Stuart-Prower-Faktor-Mangel (Faktor X-Mangel)
- Parahämophilie bzw. Owren-Syndrom oder Hypoproakzelerinämie (Faktor-V-Mangel)
- Angiohämophilie bzw. Willebrand-Jürgens-Syndrom (Fehlen oder Strukturdefekt des von-Willebrand-Faktors)
Hämophilie-Schweregrade
Hinsichtlich des Schweregrades der Hämophilie kann man konstatieren, dass die Faktor-VIII/IX-Restaktivität i.d.R. mit der Blutungsneigung korreliert. Aus diesem Grund wird die Hämophilie entsprechend der Faktoraktivität in drei Schweregrade eingeteilt:
- milde Hämophilie (Faktor VIII-/IX-Restaktivität: > 5 – 49 %): i.d.R. unauffällig, selten spontane Blutungen sowie Blutungen nach Verletzungen oder perioperativ
- mittelschwere Hämophilie (Faktor VIII-/IX-Restaktivität: 1 – 5 %): seltener spontane Blutungen sowie Blutungen nach Bagatellverletzungen oder perioperativ
- schwere Hämophilie (Faktor VIII-/IX-Restaktivität: < 1 %) mit spontanen Blutungen (v.a. Gelenke, Muskulatur) sowie seltener Hirn-, GI-Blutungen, perioperative Blutungen
Symptome der Hämophilie
Damit sich die ersten Symptome der Hämophilie zeigen muss der Faktor VIII- bzw. IX- Mangel bei < 50 % des Normalwerts liegen. Zusätzlich sind die Symptome auch abhängig von der Hämophilie-Form und/oder -Ausprägung. Zu den häufigen Symptomen bzw. Blutungen gehören:
- schmerzhafte Gelenkeinblutungen (ggf. mit dauerhaften Spätfolgen; v.a. Sprung-, Knie- und Ellenbogengelenke; häufigste Form)
- vermehrte Hämatome, v.a. an Armen und Beinen sowie am Rumpf
- verstärkte Epistaxis
- Einblutungen in Muskel- oder Fettgewebe (v.a. bedingt durch Traumata; z.B. Psoasmuskel)
- ggf. blutiges Urin (rötlich/braun)
- beim Zahnwechsel ggf. stärkere Nachblutungen
- seltener innere Blutungen (z.B. Nierenblutungen mit starken Koliken, Harnwegsverschluss durch Thromben, GI-Blutungen mit typischen Symptomen)
- Knochenblutungen (CAVE: Osteolyse durch rezidivierende Einblutungen im Knochen)
- Hirnblutungen, v.a. bei Z.n. Sturz
- bei oberflächlichen Schnittverletzungen und Schürfwunden sistieren die Blutungen entgegen der landläufigen Meinung häufig selbst
- verstärkten Regelblutungen bei betroffenen Frauen
Wie oben schon erwähnt kommt es durch die verzögerte Blutgerinnung zu ggf. immer wieder aufbrechendem Schorf und damit zu rezidivierenden Blutungen. Weiterhin ist zu berücksichtigen, dass v.a. in der Altersgruppe zw. 6 – 12 Monaten multiple Hämatome ggf. mit den Folgen von Kindesmisshandlung verwechselt werden.
Management
Initial sollte eine ausführliche, altersabhängige Blutungsanamnese erfolgen, welche sich v.a. auf Hämatomneigung, Nasen-/Zahnfleischblutung, Gelenk-/Muskelblutungen sowie bei Frauen auf Hypermenorrhoe und postpartale Blutungen konzentrieren sollte. Zusätzlich sollte eine Medikamentenanamnese erfolgen, die vor allem auf Hämophylie-typische Medikamente wie z.B. Emicizumab, Rurioctocog/Turoctocog/Damoctocog/Vonicog alfa, Eptacog beta sowie Faktor VIII/IX, Fibrinogen, Valoctocogen Roxaparvovec oder Etranacogen dezaparvovec abzielt. Auch familienanamnestisch sollten Blutungsstörungen abgeklärt werden. Während der Anamnese sollten auch relevante Differenzialdiagnosen wie das Owren- oder Rosenthal-Syndrom, der Stuart-Prower-Faktor-Mangel, das Willebrand-Jürgens-Syndrom sowie die Einnahme von Lupusantikoagulanzien oder ein Mangel an Präkallikrein oder hochmolekularem Kininogen ausgeschlossen werden.
Grundsätzlich lässt sich bei der Akutbehandlung der Hämophilie sagen, dass diese abhängig ist von der Schwere und Lokalisation der Blutung sowie ggf. auch von der zusätzlichen Einnahme von Inhibitoren im Rahmen einer dauerhaften Hämophilie-Therapie. Zu den gefährlichen Blutungsorten zählen z.B. Hirn-, gastrointestinale oder Mundbodenblutungen. Egal wie sollte ein hämostaseologisches Konzil erfolgen.
Mit der wichtigste Therapieansatz, der i.d.R. aber erst innerklinisch möglich ist, ist die Gabe des jeweils fehlenden Gerinnungsfaktors. Hierbei eignet sich die folgende Formel für die Berechnung der notwendigen Menge: 1 IE/kgKG = Gerinnungsfaktoranstieg im Plasma von 1 – 2 %. So werden bei schweren Blutungen i.d.R. 50 – 80 IE/kgKG bei Erwachsenen und 80 – 100 IE/kgKG bei Kindern verabreicht, um so einen VIII-/IX-Spiegel von 80 – 100 % zu erreichen. Entsprechend der Querschnitts-Leitlinien der BÄK zur Therapie mit Blutkomponenten und Plasmaderivaten sind die folgenden Dosierungen für die unterschiedlichen Blutungslokalisationen vorgesehen (s. Tab. 2) . Nehmen die Hämophilie A-Patient*innen z.B. eine Prophylaxe wie Emicizumab ein, soll in gleicher Art und Weise verfahren werden. Bei Hämophilie B-Patient*innen sollte unbedingt ein hämostaseologisches Konzil erfolgen. Wenn bei Patient*innen mit Inhibitoreneinahme eine lebensbedrohende Blutung besteht, sollte rekombinanter Faktor VIIa in mittlerer initialer Dosis 90 μg/kgKG oder 270 μg/kgKG als Einzeldosis verabreicht werden.
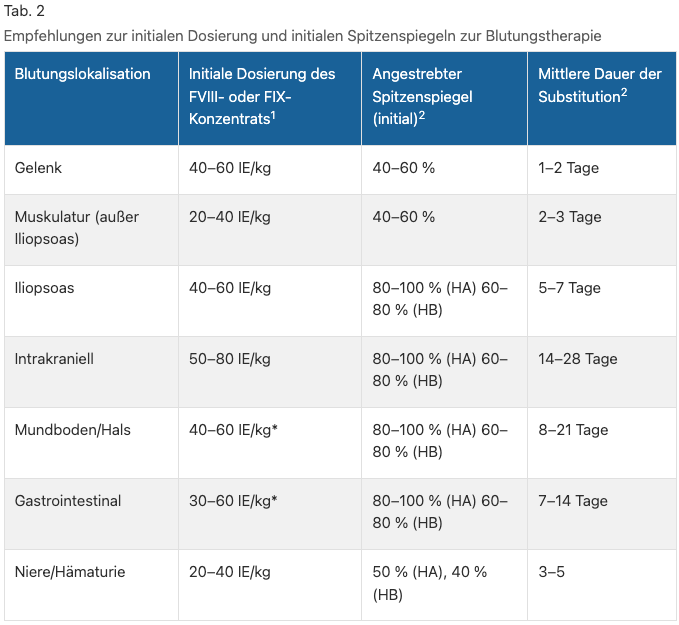
Hinsichtlich der Indikationen für die Gerinnungsfaktoren-Gabe bei Hämophilie-Patient*innen sind v.a. die folgenden Punkte zu nennen:
- Verdacht auf Gelnek-/Muskelblutung
- relevante Verletzung/Blutungen an Kopf, Hals, Mund, Ohren oder Augen
- neue oder ungewöhnliche Kopfschmerzen, insbesondere nach Trauma
- starke Schmerzen/Schwellungen und/oder starke/anhaltende Blutungen an jeder Stelle des Körpers
- offenen Wunden, die chirurgischen Verschluss, Wundkleber oder Steri-Strips erfordern
- vorausgegangenes Trauma, das zu inneren Blutungen führen könnte
- jedes invasive Verfahren oder Operationen
- gastrointestinale Blutungen
- akute Frakturen, Luxationen und Verstauchungen
- starke Menstruationsblutungen mit Gefahr von mäßiger bis schwerer Anämie oder Volumeninstabilität
Weiter kann z.B. bei schwerwiegenden Weichteilblutungen die zusätzliche Gabe von Tranexamsäure als Antifibrinolytika notwendig sein. Hierbei ist aber zu betonen, dass TXA allein nicht von Nutzen ist, jedoch einen klaren Nutzen hat bei der Kontrolle von Haut- bzw. Schleimhautblutungen sowie Menorrhagien hat (25 mg/kg i.v., max. 1,5 g bei Erwachsenen, drei- bis viermal tgl.).Zusätzlich sollte zur Behandlung des weiterhin bestehenden Blutungsrisikos nach der finalen Diagnosestellung die immunsuppressive Therapie z.B. mit Steroiden erfolgen (1 mg/kg Prednisolon, oft in Kombination mit Cyclophosphamid oder Rituximab).
Präklinisch sollte bei V.a. oder bestätigter Hämophilie der Transport in spezialisiertes Behandlungszentrum erwogen werden. Hierbei sind aber Verletzungs- bzw. Blutungsschwere zu berücksichtigen.
Hämarthrose sowie Muskel- & Weichteilblutung
- externe Kühlung mit Kühlpacks (Wechsel zw. 5 min Kühlen & 10 min Pause)
- Ruhigstellung des betroffenen Gelenks/der betroffenen Extremität
- CAVE: keine Anlage eines zirkulären Gips
- kein Röntgen indiziert, außer bei V.a. Fraktur
- Vorgehen gemäß PRICE-Schema
- P = Protection (Schutz; Ruhigstellung mit Schienung o.Ä.)
- R = Rest (Ruhe)
- I = Ice (externe Kühlung mit Kühlpacks im Wechsel zw. 5 min Kühlen & 10 min Pause)
- C = Compression (sanfter Kompressionsverband)
- E = Elevation (Hochlagerung)
Schädel-Hirn-Trauma
- Kopf-CT
- antiepileptische, medikamentöse Therapie, wenn Blutung bestätigt ist
orale Blutung
- Gabe von 5 – 10 mL TXA (500 mg / 5 ml) alle 6 h oral (CAVE: vor Schlucken 2 min im Mund behalten)
- lokale, manuelle Kompression, sofern möglich
- Kühlung mit Eis o.Ä. (Wechsel zw. 5 min Kühlen & 10 min Pause)
- ggf. Erythrozytentransfusion erwägen
urogenitale Blutung
- Flüssigkeitszufuhr erhöhen (1 Glas Wasser /h)
- Bettruhe
- bei ausbleibender Besserung innerhalb von 24 h Faktorersatz durchführen
- CAVE: Tranexamsäure kontraindiziert
Schmerzen bei Hämophilie
- Gabe von COX-2-Hemmer (z.B. Celecoxib, Meloxicam) aufgrund des besseren Wirkungsprofils
- ggf. zusätzlich Opiate (z.B. Tramadol) und/oder Paracetamol erwägen
weitere Besonderheiten beim Management
- Mobilisierung nur mit Bedacht
- nur Anlage von pVK (CAVE: Blutungs-/Einblutungsgefahr bei ZVK, i.m.-Applikation etc.)
- Anlage von Kompressionsverbänden an Punktionsstellen
- keine rektale Temperaturmessung
- keine Plexusblockade ohne Substituierung
- bei Koronarangiographie vorher Faktorausgleich, auch im Notfall
- CAVE bei ETI: Risikos von bukko-pharyngealen Hämatomen
Exkurs – Millionentherapie bei Hämophilie B
2022/2023 wurde das Medikament HEMGENIX® der Firma CSL Behring von der FDA für die USA sowie von der EMA für EU zugelassen und brach mit einigen langbestehenden Grundsätzen in der Therapie der Hämophilie B. HEMGENIX® (Wirkstoffname: Etranacogene Dezaparvovec) ist eine Gentherapie, die die Gerinnungsfaktoren-Substitution bei erwachsenen Patienten mit schwerer und mittelschwerer Hämophilie B ohne Faktor-IX-Inhibitoren in der Vorgeschichte lebenslang bzw. für einen sehr langen Zeitraum ersetzen kann. In der Zulassungsstudie HOPE-B konnte gezeigt werden, dass 96 % aller Patienten nach der Infusionstherapie keine regelmäßige Prophylaxe mit Faktor-IX mehr benötigten und sich die jährl. Blutungsrate um 64 % verringerte.
Die Wirkweise von HEMGENIX® sieht wie folgt aus: Als Wirkstoff dient ein Adeno-assoziiertes Virus vom Serotyp 5 (AAV5), welches vermehrungsunfähig ist. Der AAV-Vektor enthält den Bauplan für die Bildung des Faktor-IX-Proteins in Form eines Gens und überträgt diesen auf ein paar körpereigene Leberzellen der betroffenen Personen. So ist es mit der einmaliger Applikation möglich, dass die Leberzellen so dauerhaft Faktor-IX-Protein herstellen, die sogar 5 bis 8 mal aktiver sind als normal. Damit der AAV ganz spezfisch wirkt enthält der eine besondere, angepasste Version der menschlichen Gerinnungsfaktor IX-Genvariante R338L sowie zusätzlich den LP1 (leberspezifischer Promotor), durch welchen HEMGENIX® genau an den Leberzellen andockt.
Die einmalige Gabe erfolgt als Einzeldosis-Infusion i.v. mit einer Laufrate von 8 mL/h. Hierbei wird die empfohlene Dosis von 2 x 1013 Genomkopien/kgKG (= 2 mL/kgKG) mit 9 mg/mL NaCl 0,9% verdünnt. Eine Push-Injektion oder Bolusinfusion ist kontraindiziert. Nach der Applikation kann es jedoch mehrere Wochen dauern bis die therapeutische Wirkung merkbar ist (ggf. ist auch die zusätzliche Gabe von exogenem humanem Faktor IX notwendig).
HEMGENIX® hat, wie schon einige andere Medikamente zuvor, große Kritik ausgelöst, da es einen ggf. fragwürdigen Rang als das aktuell teuerste Medikament der Welt eingenommen hat. CSL Behring hat den Handelspreis von HEMGENIX® auf 3.500.000 $ festgelegt. Den hohen Preis begründet CSL Behring mit der teuren und langwierigen Entwicklung sowie dem kleinen Patientenkollektiv für welches die Therapie infrage kommt. Zusätzlich wird argumentiert, dass die hohen Einmalkosten für HEMGENIX® sich nach ca. 10 Jahren im Vergleich zur regelmäßigen Prophylaxe mit Gerinnungsfaktoren amortisiert haben (Berechnung von CSL Behring ergaben Einsparungen von 5 – 5,8 Millionen US-$ pro Patient). Hier gilt es aber auch zu betonen, dass es in einigen Fällen dazu kam, dass die Wirkung von Etranacogene Dezaparvovec verflog und eine zweite Gabe von HEMGENIX® aktuell nicht vorgesehen ist bzw. nicht erprobt/überprüft ist. Kritiker wie das US-amerikanische Institute for Clinical and Economic Review sehen insgesamt den Preis für die Gentherapie als zu hoch angesetzt an und haben in ihrer Analyse einen fairen Preis von < 3.000.000 $ ermittelt.
Quellen
- Australian Haemophilia Centre Directors’ Organisation, und National Blood Authority, Hrsg. „Guidelines for the management of haemophilia in Australia“. National Blood Authority, 2016. https://blood.gov.au/system/files/HaemophiliaGuidelines-interactive-updated-260317v2.pdf.
- Barthels, Monika, Cornelia Wermes, Wolfgang Voerkel, und Christoph Bidlingmaier. „DHG e.V.:Hämophilie“. Deutsche Hämophiliegesellschaft e.V. Deutsche Hämophiliegesellschaft zur Bekämpfung von Blutungskrankheiten e.V. (DHG e.V.), 16. Juni 2020. https://www.dhg.de/blutungskrankheiten/haemophilie.html.
- Bayer Vital GmbH. „Hämophilie erkennen“. Faktorviii – Bayer Hämophlie. Zugegriffen 11. April 2024. https://www.faktorviii.de/fuer-patienten/haemophilie/symptome.
- Bleich, Markus, Andreas Draguhn, Heimo Ehmke, und Dominique Singer. Physiologie. Herausgegeben von Hans-Christian Pape, Armin Kurtz, Stefan Silbernagl, Rainer Klinke, Rüdiger Gay, und Astried Rothenburger. 10., Vollständig überarbeitete Auflage. Stuttgart New York: Georg Thieme Verlag, 2023. https://doi.org/10.1055/b000000639.
- Bündnis zur Förderung der Sicherheit von Hämophilen e.V. „Notfallbehandlung von Patienten mit einer Gerinnungsstörung: Empfehlungen für Ärzte“. BFSH e.V. – Bündnis zur Förderung der Sicherheit von Hämophilen e.V., 1. März 2022. https://www.bfsh.info/wp-content/uploads/2022/03/BFSH_Notfallempfehlungs.pdf.
- Centers for Disease Control and Prevention. „Data & Statistics | Hemophilia“, 12. Juli 2023. https://www.cdc.gov/ncbddd/hemophilia/data.html.
- CSL Behring. „Gentherapie für Hämophilie B“, 2. Mai 2023. https://www.cslbehring.de/news/2023/launch-gentherapie-hemophilie-b.
- DAP Networks GmbH. „HEMGENIX® erhält bedingte Zulassung durch die Europäische Kommission“. DeutschesApothekenPortal, 7. März 2023. https://www.deutschesapothekenportal.de/rezept-retax/nachrichten/arzneimittel/detail/?cHash=694fce566a228e12ac5eb71f07945e7a&tx_news_pi1%5Baction%5D=detail&tx_news_pi1%5Bcontroller%5D=News&tx_news_pi1%5Bnews%5D=5756.
- Dunst, Gregory, Frank Antwerpes, Fiona Walter, Matthias Firner, Andreas Hecker, Philip Denz, Lisa Schönwälder, u. a. „Hämophilie“. DocCheck Flexikon, 21. März 2024. https://flexikon.doccheck.com/de/H%C3%A4mophilie.
- Goudemand, Jenny, Anne Lienhart, Sandrine Meunier, Thierry Lambert, Catherine Ternisien, Gaële Comte, und Gilles Bagou. „Notfall-Leitlinie Hämophilie“. Orphanet Deutschland, 6. Oktober 2009. https://www.orpha.net/pdfs/data/patho/Emg/de/Emergency_Haemophilie-dePro646.pdf.
- Grisoli, Anne. „Therapeutics: Hemophilia Management in the ED“. Taming the SRU, 16. Februar 2022. https://www.tamingthesru.com/blog/therapeutics/hemophilia.
- Haefely, Andrea. „Eine Spritze für 3,5 Mio. Dollar: 7 Antworten zum teuersten Medikament der Welt“. Beobachter, 1. Dezember 2022. https://www.beobachter.ch/gesundheit/medizin-krankheit/7-antworten-zu-hemgenix-dem-teuersten-medikament-der-welt-551621.
- „Hämophilie“. In Wikipedia, 9. April 2024. https://de.wikipedia.org/w/index.php?title=H%C3%A4mophilie&oldid=243899129.
- Hanley, John, Mary Mathias, Emma Franklin, Chris Harrington, Oliver Chapman, Kate Talks, und Stephanie Smith. „Emergency and out of Hours Care for Patients with Bleeding Disorders – Standards of Care for Assessment and Treatment.“, 2009.
- „Hemophilia: Types, Causes, Symptoms, and Treatment“. Pfizer. Zugegriffen 11. April 2024. https://www.pfizer.com/disease-and-conditions/hemophilia.
- Holstein, Katharina. „Angeborene und erworbene Hämophilie – DGIM Innere Medizin – eMedpedia“. Springer Medizin – e.Medpedia, 20. April 2023. https://www.springermedizin.de/emedpedia/detail/dgim-innere-medizin/angeborene-und-erworbene-haemophilie?epediaDoi=10.1007/978-3-642-54676-1_94.
- Kääb, Georg. „Das teuerste Medikament der Welt kommt aus den Niederlanden“. |transkript, 28. November 2022. https://transkript.de/artikel/2022/das-teuerste-medikament-der-welt-kommt-aus-den-niederlanden/.
- Mahlangu, Johnny N., Anne Gilham, und Medical and Scientific Advisory Council of the South African Haemophilia Foundation. „Guideline for the Treatment of Haemophilia in South Africa“. South African Medical Journal = Suid-Afrikaanse Tydskrif Vir Geneeskunde 98, Nr. 2 Pt 2 (Februar 2008): 126–40.
- Maucher, Isabelle Viktoria. „Neueinführung Hemgenix bei Hämophilie B“. Gelbe Liste, 26. April 2023. https://www.gelbe-liste.de/neue-medikamente/hemgenix-haemophilie-b.
- Naddaf, Miryam. „Hämophilie: Das 3,5-Millionen-Dollar-Medikament“. Spektrum.de, 20. Dezember 2022. https://www.spektrum.de/news/haemophilie-das-3-5-millionen-dollar-medikament/2091675.
- ———. „Researchers Welcome $3.5-Million Haemophilia Gene Therapy — but Questions Remain“. Nature 612, Nr. 7940 (6. Dezember 2022): 388–89. https://doi.org/10.1038/d41586-022-04327-7.
- National Bleeding Disorders Foundation. „Hemophilia Fast Facts“. Zugegriffen 12. April 2024. https://www.hemophilia.org/bleeding-disorders-a-z/overview/fast-facts.
- Paul-Ehrlich-Institut. „Meldungen – Erstes Gentherapeutikum gegen Hämophilie B erhält Zulassungsempfehlung“, 20. Februar 2023. https://www.pei.de/DE/newsroom/hp-meldungen/2022/221220-gentherapeutikum-haemophilie-b.html.
- Paul-Ehrlich-Institut – Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel. „Welttag der Hämophilie: Therapiemöglichkeiten der Bluterkrankheit“, 17. April 2023. https://www.pei.de/DE/newsroom/hp-meldungen/2023/230414-welttag-haemophilie.html.
- Pfizermed AT. „Hämophilie | Seltene Erkrankungen“, 1. Februar 2024. https://www.pfizermed.at/therapiegebiete/seltene-erkrankungen/hämophilie.
- Schlenkrich, Uwe. „Gerinnungspräparate“. Interessengemeinschaft Hämophiler e.V. Zugegriffen 15. April 2024. https://www.igh.info/medikamentenliste/medikamentenliste/gerinnungspraeparate.html.
- Srivastava, Alok, Elena Santagostino, Alison Dougall, Steve Kitchen, Megan Sutherland, Steven W. Pipe, Manuel Carcao, u. a. „WFH Guidelines for the Management of Hemophilia, 3rd Edition“. Haemophilia 26, Nr. S6 (August 2020): 1–158. https://doi.org/10.1111/hae.14046.
- Streiff, Michael. „Hämophilie – Hämatologie und Onkologie“. MSD Manual Profi-Ausgabe, 1. September 2023. https://www.msdmanuals.com/de-de/profi/h%C3%A4matologie-und-onkologie/gerinnungsst%C3%B6rungen/h%C3%A4mophilie.
- Takeda Pharma Vertrieb GmbH & Co. KG. „Notfallmaßnahmen bei Hämophilie“. myHaemophilie (blog). Zugegriffen 11. April 2024. https://www.myhaemophilie.org/leben/berufseinsteiger/notfall.
- Tallen, Gesche. „Häufigkeit: Wie oft kommt eine Hämophilie vor?“ Kinderblutkrankheiten – Informationsportal zu Blut- und Gerinnungserkrankungen bei Kindern und Jugendlichen, 22. August 2022. https://www.kinderblutkrankheiten.de/erkrankungen/gerinnungsstoerungen/haemophilie/haeufigkeit/.
- The Royal Children’s Hospital Melbourne. „Clinical Practice Guidelines: Haemophilia“, 1. Mai 2023. https://www.rch.org.au/clinicalguide/guideline_index/Haemophilia/.
- Thieme – TRIAS. „Welttag der Hämophilie – TRIAS Verlag – Gesundheit“. Zugegriffen 11. April 2024. https://www.thieme.de/de/gesundheit/welttag-haemophilie-37731.htm.
- World Federation of Hemophilia. „Hemophilia“. WFH eLearning Platform (blog). Zugegriffen 11. April 2024. https://elearning.wfh.org/elearning-centres/hemophilia/.
Sei der Erste der einen Kommentar abgibt